AI 赋能抗微生物耐药性预测:全基因组测序助力药敏检测提速

面对日益严峻的"超级细菌"挑战,中国医学科学院北京协和医院检验科杨启文教授团队与杭州杰毅生物技术有限公司等机构的研究团队取得了突破性进展。研究者利用机器学习和大规模全基因组测序数据,开发出能够精准预测肺炎克雷伯菌耐药表型的模型,将传统需要 48 至 72 小时的药敏检测流程,缩短至 24 至 30 小时(基于分离株测序)或 24 小时以内(基于血培养富集测序),为临床早期精准用药争取了关键时间窗口。
破解"时间差"难题:从基因型到表型的智能跨越
肺炎克雷伯菌是引起医院内感染的常见病原体,其多重耐药特性常导致临床治疗陷入困境。目前临床上的"金标准"是传统的培养法药敏试验,但整个过程需耗时 48 至 72 小时,期间临床医生常需进行经验性用药,可能造成治疗延迟或抗菌药物不合理使用。
虽然全基因组测序能快速获取细菌的全部遗传信息,但基因与最终耐药表现之间并非简单的一一对应关系——一个基因可能影响多种药物,而一种耐药性也可能由多个基因共同决定。这种复杂性成为了精准抗微生物治疗的"瓶颈"。
为了破解这一难题,研究人员构建了基于机器学习算法的预测模型。研究者收集了来自中国、欧洲、美洲和亚洲的 5239 株肺炎克雷伯菌的基因组及药敏数据,时间跨度近 20 年(2004-2022 年),构建了该领域迄今为止规模最大的数据集,覆盖了 11 种临床常用抗生素。
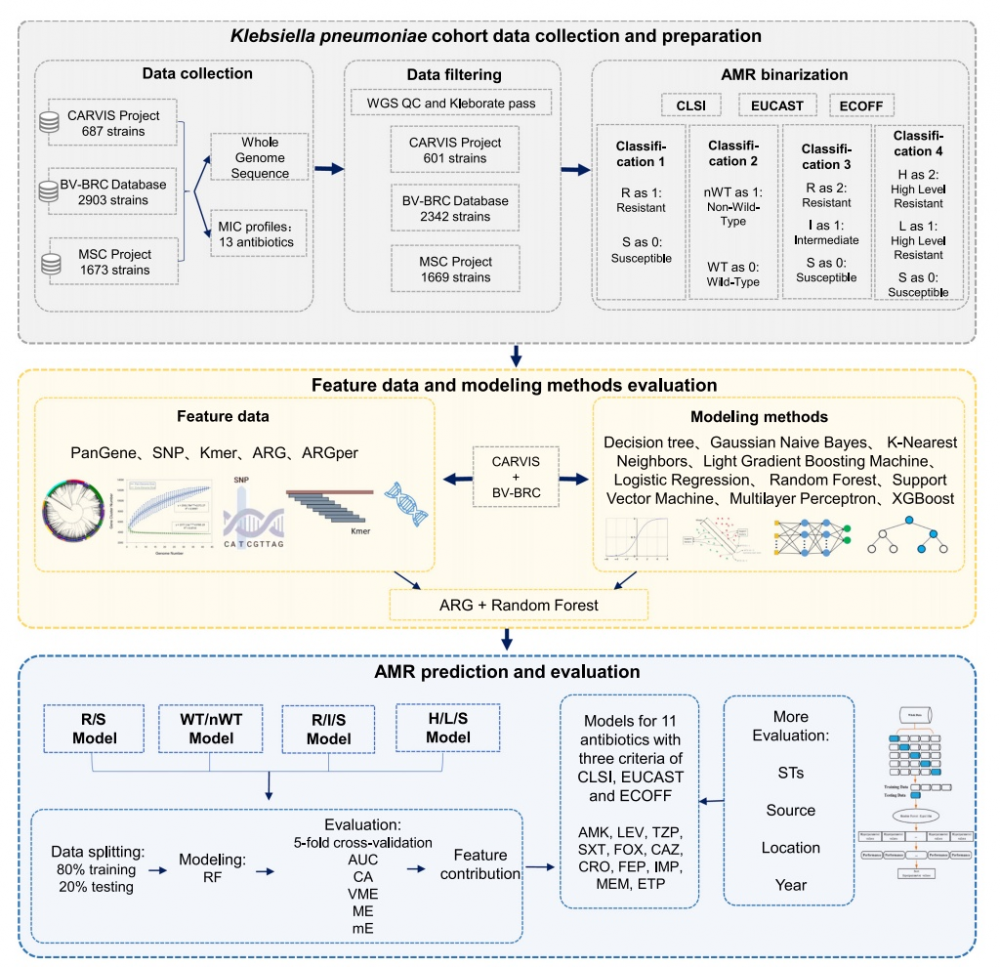
多层级耐药预测:从二元分类到更细分层
这个模型的最大突破在于它能够实现多层级的耐药表型预测,为临床决策提供比"敏感(S)/耐药(R)"二元区分更精细的指引:
1. 区分敏感、中介与耐药(R/I/S): 准确分辨出"中介"这一临床上的灰色地带,为医生调整给药方案或剂量提供参考依据。
2. 区分高水平耐药与低水平耐药(H/L): 精准判断耐药的程度。高水平耐药通常意味着标准用药方案将完全无效,而低水平耐药或许可通过调整给药方案加以应对。
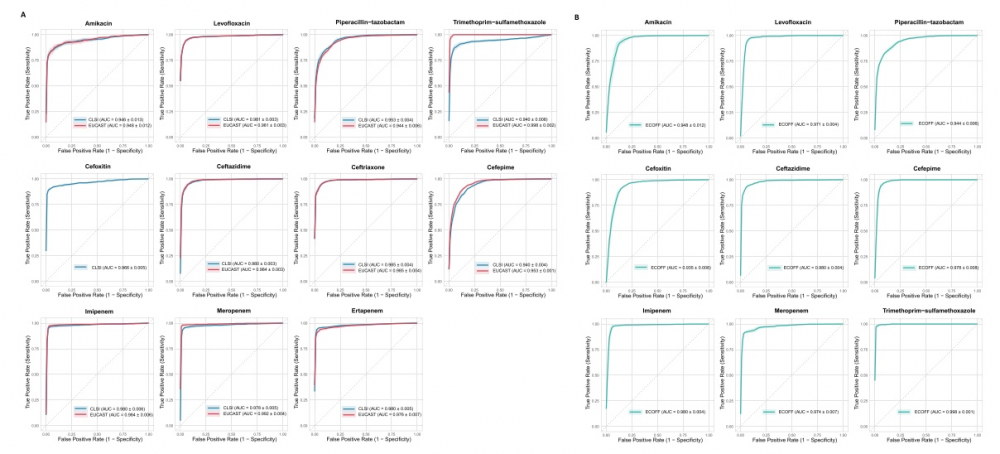
在区分敏感和耐药(R/S)时,该模型对所有 11 种测试抗生素的受试者工作特征曲线下面积(AUC)均值超过 0.9,平均分类一致率(CA)达 0.96,展现出极佳的判别能力。在区分耐药、中介和敏感(R/I/S)模型中,整体性能同样出色,平均 AUC 与 CA 均大于 0.9,其中平均极重大误差(即耐药株被误判为敏感株的比例)为 1.3%,平均重大误差(即敏感株被误判为耐药株的比例)为 1.5%,表明模型将耐药株误判为敏感株的风险极低,为临床安全应用提供了保障。
解码耐药地图:模型关注到了哪些关键基因?
研究的精准性得益于机器学习中的随机森林算法能够评估每个耐药基因对预测模型的贡献度(以 Gini 指数衡量)。该研究发布了不同抗生素的"十大关键耐药基因"列表,进一步验证了模型的生物学可解释性。
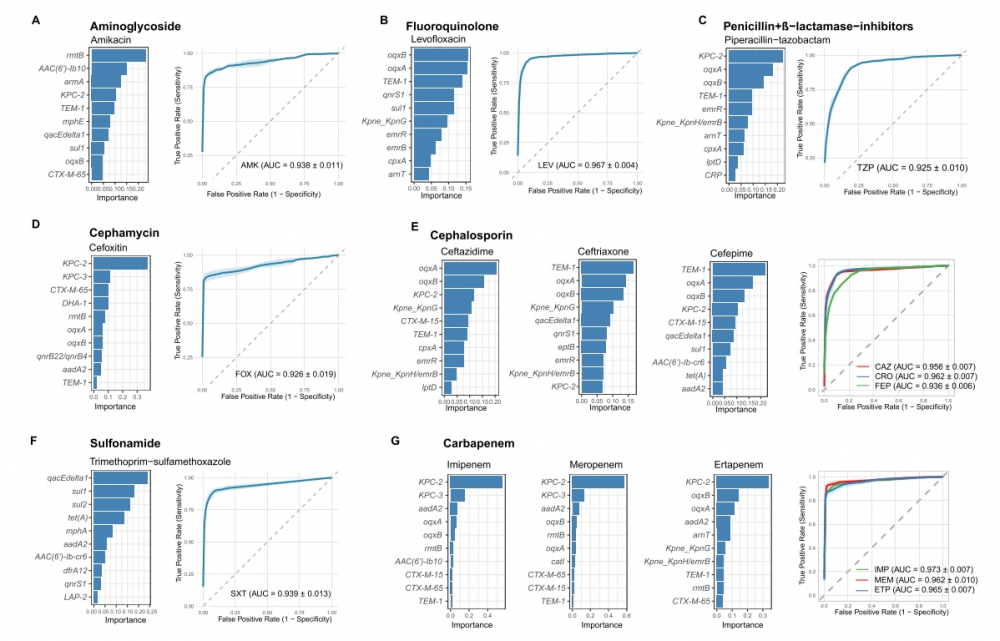
例如,碳青霉烯酶基因 KPC-2 是模型预测碳青霉烯类药物(如亚胺培南、美罗培南)和广谱头孢菌素耐药的首要依据;超广谱β-内酰胺酶基因 CTX-M-15 则与第三代头孢菌素耐药密切相关。这些计算得出的关键特征,与已知的耐药生化机制高度吻合,证明了模型的科学可靠性。此外,模型还揭示了一些偏离经典机制预期的关联,如 ompK36/37 等孔蛋白基因与左氧氟沙星耐药表型的强相关,为后续耐药机制研究提供了新线索。
跨地域、跨时间、跨菌株的稳健表现
一个优秀的临床预测工具必须具备广泛的适用性。研究人员对模型进行了严苛的"压力测试",为模型的泛化能力提供了坚实证据。
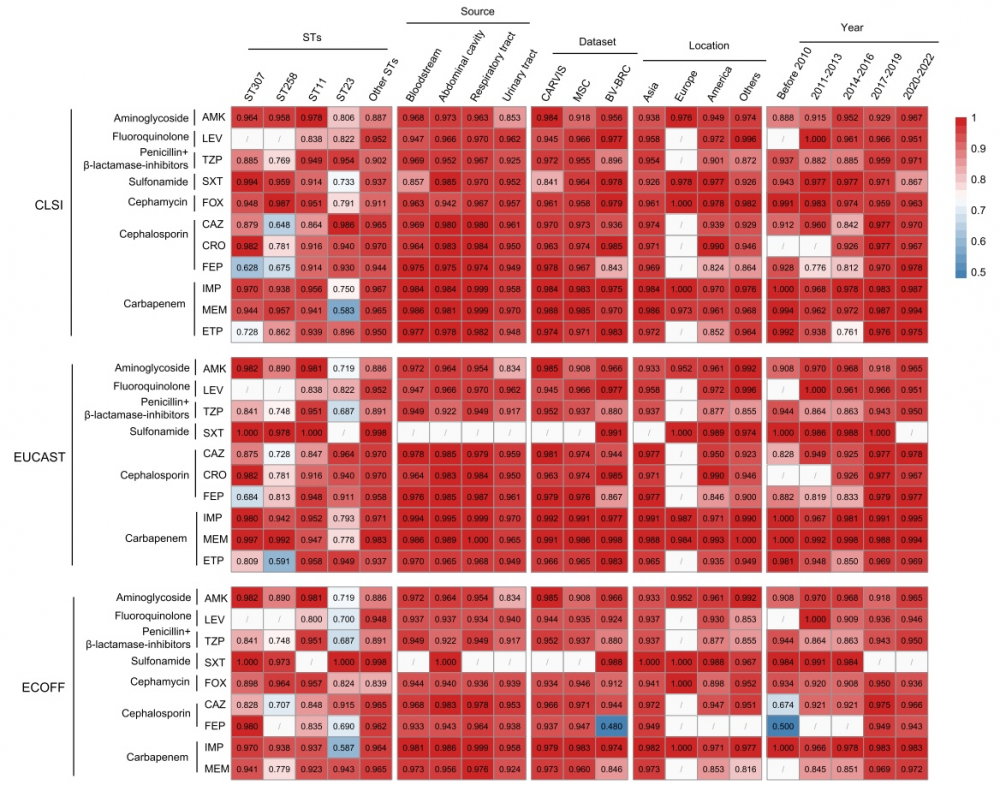
跨地域和人群:当用亚洲、欧洲、美洲等不同地区的菌株分别验证时,模型预测的 AUC 绝大部分维持在 0.8 以上,多数超过 0.9。
跨年份、克隆变迁:将 2004 年至 2022 年的菌株按时间段(2010 年以前、2011–2013 年、2014–2016 年、2017–2019 年、2020–2022 年五个时段)划分后评估,模型对各年份段的预测能力均保持稳健,表明其不受短期流行克隆变迁的显著影响。
不同感染部位: 无论细菌是从血液、呼吸道还是泌尿道分离得来,模型的性能表现基本一致。
高危耐药克隆:即使面对 ST11、ST258、ST307 这类公认的高危多重耐药克隆群,模型的预测效能依然稳健。
临床转化:血培养富集测序 24 小时内可获得药敏预测
研究人员利用 34 例临床血培养阳性标本进行了验证,采用血培养富集测序路径,从血培养阳性报警到获得全基因组测序药敏预测结果控制在 24 小时以内,成本约 85 元人民币。验证结果显示,预测准确率对于多数抗生素达 90% 以上,其中厄他培南的敏感/耐药预测准确率达到 100%。
相较于传统培养法需 48 至 72 小时,这一速度显著缩短了报告周转时间,使临床医生可以在感染早期即获得全面的耐药谱信息,从而在治疗"黄金窗口期"实施精准靶向治疗。
研究团队指出,该模型的设计已考虑到未来在临床实验室中的部署,可作为轻量软件模块无缝整合进现有的测序分析系统,并输出包含置信度评分的标准化耐药报告,便于与医院信息系统对接。
未来展望
尽管成果显著,研究人员也客观指出了当前模型的局限性:对于部分抗生素的"中介"或"低水平耐药"类别,预测性能相对较弱,这主要是因为在真实临床数据中该类别的样本天然稀缺。此外,细菌的耐药性是一个动态演化的过程,若仅采用静态模型而不进行持续更新,可能无法识别新出现的耐药决定因子,导致虚假的敏感预测结果。
为此,研究团队展望了一个"持续学习框架",计划通过不断纳入涵盖新耐药机制的全球性数据,定期对模型进行更新和再验证,以保持其长期的临床有效性和普适性。
这项研究不仅标志着我国在抗微生物耐药领域的精准诊断技术取得重大进展,也为最终实现个体化抗感染治疗提供了强有力的新工具。
杰毅生物耐药基因设计思路和优势
Q-mNGS®、SeqPlex®tNGS 耐药基因覆盖度
- 覆盖常见病原体的 60 种常见耐药基因
- 78 种毒力因子
设计思路
- 有据可查:基于 PubMed 高引用研究成果。
- 专家认可:复旦大学附属华山医院抗生素研究所专家审核。
- 知识拓展:分析 1 万+株 ESKAP 细菌的基因与 MIC 表型相关性,支持表型预测。
设计亮点
- 关键分型覆盖:包括 KPC-3 及其亚型等重要耐药分型。
- 罕见基因检测:覆盖数据库支持的少见耐药基因,并提供统计相关性数据。
- 全面注释:涵盖所有抗生素类别,支持 ESKAP 细菌表型预测注释。
