近日,广东省临床检验中心公布了广东省 2025 年下呼吸道感染、血液微生物 cfDNA、中枢神经系统感染宏基因组高通量测序室间质评结果。杰毅生物旗下杭州杰毅医学检验实验室以高分成绩顺利通过以上三项室间质评!
01. 下呼吸道感染病原体 mNGS 室间质评
本次预研活动从检测分析敏感性、特异性、重复性及稳健性等多个方面考核了省内临床实验室应用 mNGS 技术检测下呼吸道标本中病原微生物的能力。总体而言,各实验室的检测流程差异显著,对多数常见病原体的检测能力较好,但对特殊结构微生物的检出能力、生信分析精准度及流程稳定性仍需重点提升。
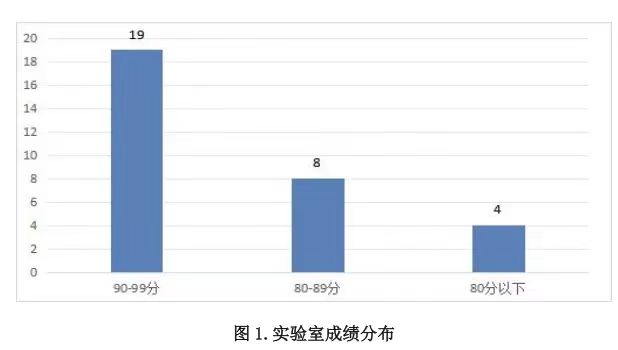
本次接受报名的实验室 33 家,实际收到 31 家有效回报结果。如下图所示,31 家实验室所得成绩分布如下:
- 90~99 分(含 90 分)的实验室 19 家;
- 80~89 分(含 80 分)的实验室 8 家;
- 低于 80 分(不含 80 分)的实验室 4 家。
按照≥80 分为合格的标准,合格率为 87.0%(27/31)。
02. 血液微生物 cfDNA mNGS 室间质评
本次预研活动从检测结果的准确性、稳健性、重复性以及报告解读等多个方面考核了 mNGS 实验室检测血浆微生物 cfDNA 诊断感染性疾病的能力。各实验室的检测流程差异较大,整体检测能力较好,但不同实验室间差异能力较大,菌株鉴定能力仍需提高。
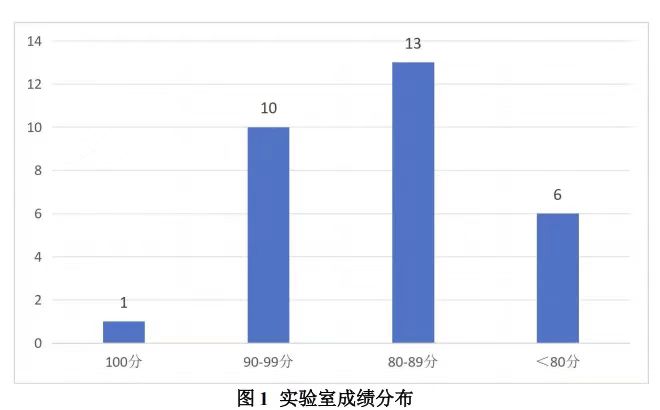
本次接受报名的实验室共 32 家,实际收到 30 家有效回报结果。如下图所示,基于评分方法,30 家实验室所得成绩分布如下:
- 100 分的实验室 1 家;
- 90~99 分(含 90 分)的实验室 10 家;
- 90~80 实验室有 13 家;
- 低于 80 分的实验室共 6 家。
按照≥80 分为合格的标准,合格率为 80%(24/30)。
03. 中枢神经系统感染 mNGS室间质评
本次预研活动从检测分析敏感性、特异性、重复性及稳健性等多个方面考核了省内临床实验室应用 mNGS 技术检测中枢神经系统感染(脑脊液)病原体的能力。总体而言,各实验室对多数常见细菌、真菌及 DNA 病毒的检测能力较好,总体合格率达 100%,但对低载量 RNA 病毒的敏感性、背景污染的控制能力及检测流程的稳定性仍需重点提升。
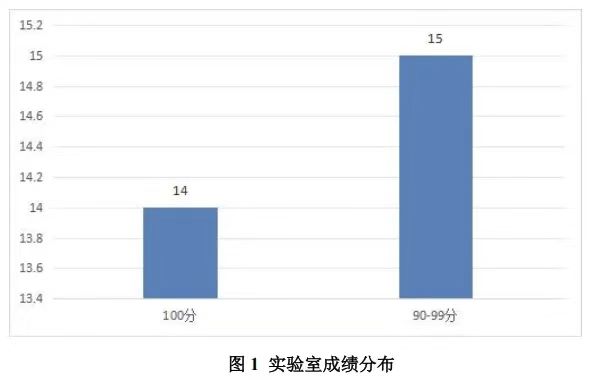
本次接受报名的实验室 31 家,实际收到 29 家有效回报结果。如下图所示,基于评分方法,29 家实验室所得成绩分布如下:
- 100 分的实验室 14 家;
- 90~99 分(含 90 分)的实验室 15 家。
按照≥90 分为合格的标准,合格率为 100%(29/29)。
聚焦临床疑难、复杂感染患者病原诊断的难题与挑战,杰毅生物持续推动 mNGS 检测技术及应用的标准化升级,公司自 2020 年推出的 Q-mNGS™定量宏基因组检测,经过技术优化升级,实现病原体检测性能的多维度提升:1. 通过正向广谱富集技术,高效富集血浆样本中低载量病原体核酸,显著提升检测灵敏度。2.NGSmaster™全自动一站式建库仪采用 PCR-free 建库方案,从实验流程中最大程度降低病原体核酸污染风险。
3. 覆盖 35257 种病原体及 60 种耐药基因、78 种毒力基因的高质量数据库,确保罕见病原体检出,CNV 分析特异性提示肿瘤信号,提升感染、肿瘤鉴别诊断效能。
4. 基于 50w 检测大数据,建立病原体分级、分类汇报原则,结合 AI 智能化生信分析,13 小时完成报告,实现病原体精准、快速鉴别,让 mNGS 检测更加契合临床所需。
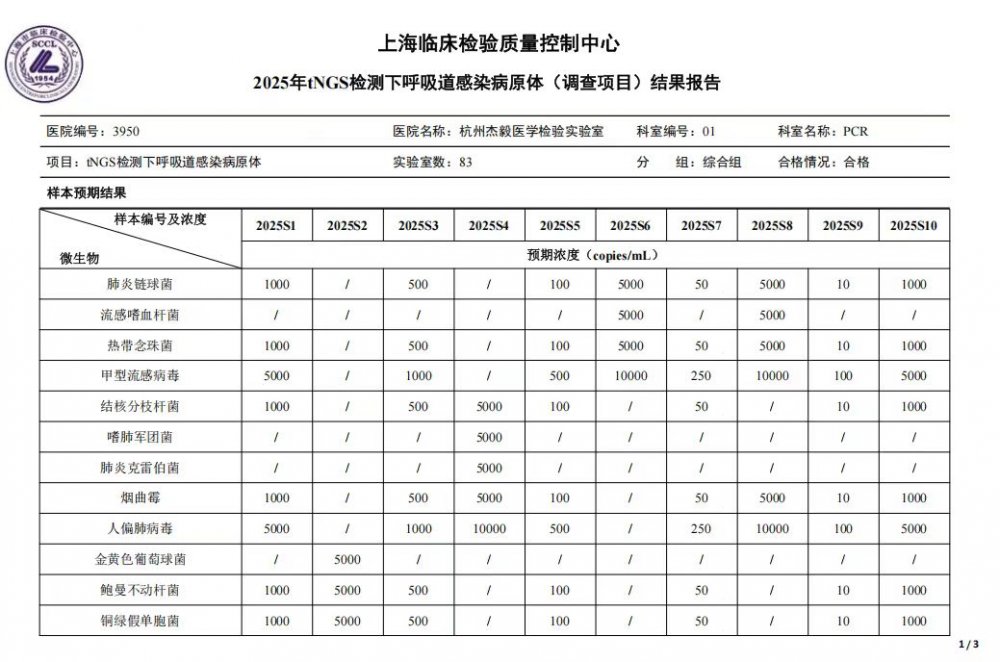
凭借高度自动化的检测平台与严格的实验室质量管理体系,杰毅医学检验实验室自 2022 年起已连续 3 年以优异成绩通过国家卫生健康委临床检验中心组织的全国下呼吸道感染、血液微生物 cfDNA、中枢神经系统感染宏基因组高通量测序等项目以及上海临床检验质量控制中心组织的血浆微生物游离 DNA 基因组高通量测序室间质评等项目,充分验证了杰毅生物 Q-mNGS 技术方案及检测能力的专业度。每一份室间质评高分成绩,既是认可,更是我们持续精进的责任。杰毅生物始终坚守初心和愿景——让每位感染患者第一时间得到精准诊疗。未来,我们将继续追求更专业、更精准、更可靠的检测能力,致力于成为感染精准诊断领域最值得信赖的合作伙伴。